CueMol is a free computer program for the macromolecular structure visualization. It aims to visualize the crystallographic models of macromolecules with the user-friendly interfaces. Currently supported files are molecular coordinates (PDB format), electron density (CCP4, CNS , and BRIX formats), MSMS surface data, and APBS electrostatic potential map.
Main functions
Display proteins and nucleic acids
Supported input format is PDB (molecular coordinates).
- simple stick model
- main-chain trace (Calpha backbone, phosphate backbone, …)
- ball-and-stick model
- CPK model
Functions as a molecular viewer
- [advt]Residue selection by clicking
- Residue selection by chain and residue numbers
- Change or create new display for the selected residues
- Display symmetric molecules
- Display unit cell
- Hardware stereo support
- Display the distance between atoms
- Dump/restore the current state to/from the file (workspace)
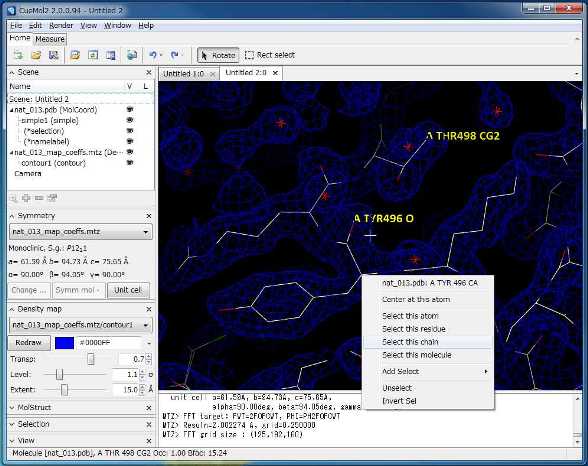
Display electron-density maps
Supported input formats are CCP4 (type2), X-PLOR/CNS (ascii), and BRIX
- Mesh and surface display of electron-density maps (you can change the contour level dynamically!!)
- Display electron density of an arbitrary (cubic) region.
- Geometric object (solid/dashed lines)
- Molecular surface object (generated by MSMS program)

Be the first to comment